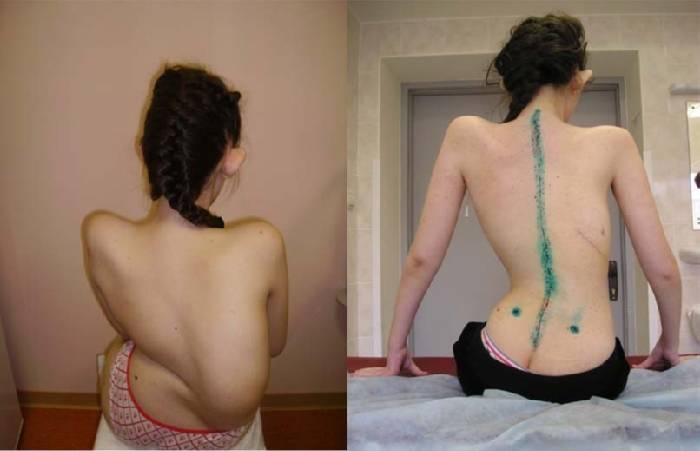
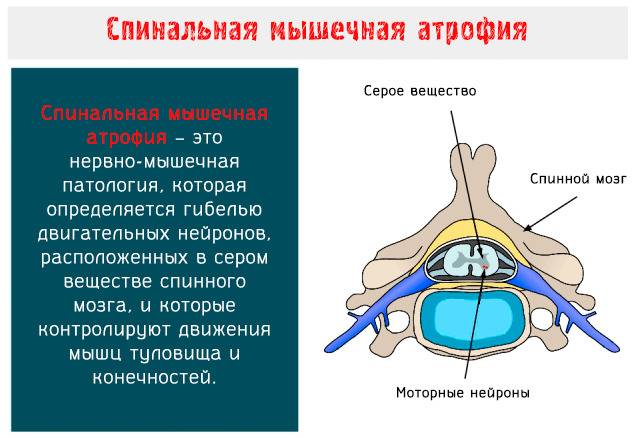
Клинические проявления
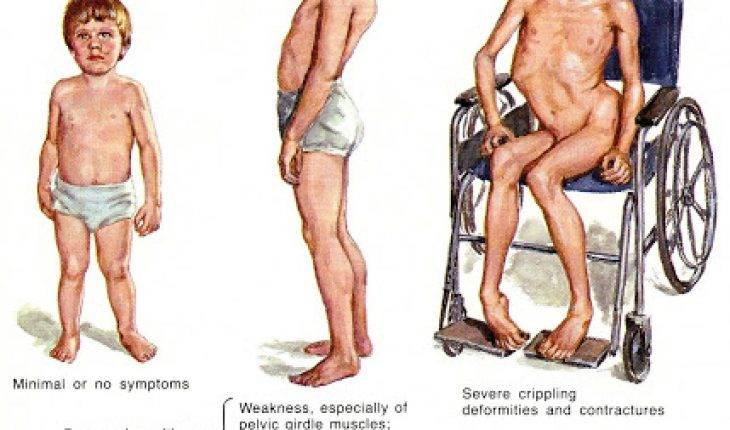
Клинические проявления весьма разнообразны по тяжести и течению. Наряду с тяжелыми формами болезни, протекающими с развитием мышечной слабости и расстройствами дыхания вскоре после рождения, с нарушениями, приводящими к смерти в первые годы жизни, имеются относительно доброкачественные формы, при которых больные доживают до юношеского или зрелого возраста.
Различают раннюю детскую (врожденную), а также детскую и поздние формы заболевания.
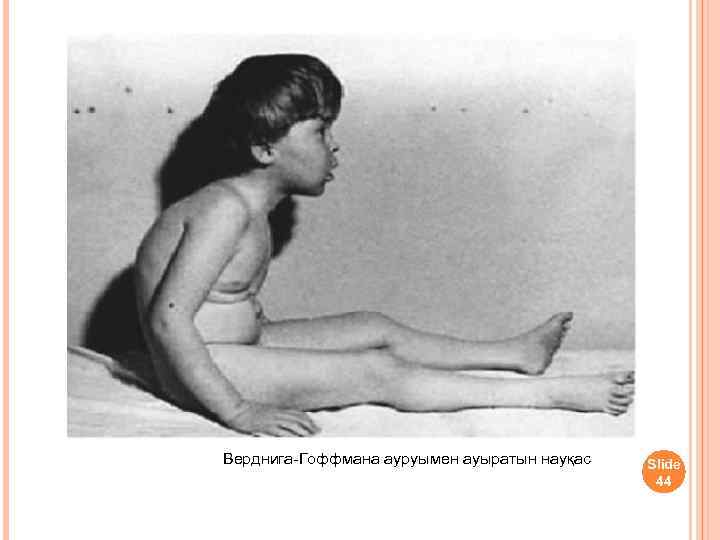
Ранняя детская форма амиотрофии клинически может проявляться во внутриутробном периоде или на первом году жизни. Отмечается позднее и вялое шевеление плода. Ребенок рождается с двигательными нарушениями, почти полным отсутствием движений в первые месяцы жизни. При обследовании обнаруживается отсутствие или снижение сухожильных рефлексов, атрофия мышц, разболтанность суставов. Врожденная миатония (см.) описана Оппенгеймом (Н. Oppenheim, 1900). По мнению автора, считавшего ее самостоятельной болезнью, в отличие от амиотрофии Верднига—Гоффманна, при этой форме не отмечается прогрессирования. Ведущим симптомом является атония мышц, мышечная слабость в проксимальных отделах конечностей и туловища, сухожильные рефлексы снижены. Однако многие современные авторы полагают, что врожденная миатония является доброкачественным вариантом ранней формы амиотрофии Верднига—Гоффманна. При ранней детской форме продолжительность жизни от 1 до 7 лет.
Детская форма амиотрофии начинается в возрасте до 4 лет, отличается от первой более медленным течением. Болезнь носит прогрессирующий характер. Длительность заболевания различна. Летальный исход наступает обычно до 14 лет.
Поздние формы спинальной амиотрофии: ювенильная форма амиотрофии и форма с более поздним началом — амиотрофия Кугельберга—Веландера. Описаны Кугельбергом и Веландером (Е. Kugelberg, L. Welander, 1954).
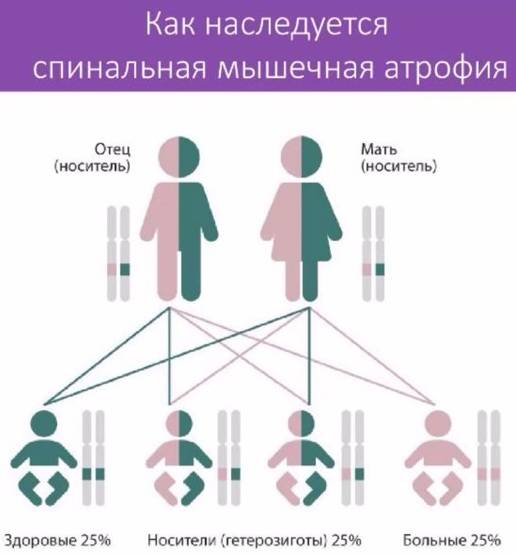
Заболевание характеризуется медленно прогрессирующей мышечной слабостью, атрофией мышц, наличием фасцикуляций, отсутствием пирамидных симптомов. Передается по аутосомно-рецессивному типу. Встречается у мужчин в 2 раза чаще, чем у женщин. У большинства больных наблюдается слабость проксимальных отделов верхних и нижних конечностей. Атрофия мышц, возникающая во всех случаях, может быть завуалирована наличием жировой клетчатки, часто обнаруживается гипертрофия ягодичных и икроножных мышц. Заболевание медленно прогрессирует, больные живут в среднем до 40 лет, иногда дольше. В более поздних стадиях в патологический процесс вовлекается мускулатура дистальных отделов конечностей, однако течение заболевания доброкачественное и в поздних его стадиях двигательные функции относительно сохранены, больные способны передвигаться, обслуживать себя.
Биохимические исследования
При амиотрофии установлено умеренное повышение активности ферментов: альдолазы, креатинфосфокиназы, трансаминаз в сыворотке крови. Однако по сравнению с первичными миопатиями (см.) это повышение выражено значительно слабее. Другим характерным биохимическим признаком амиотрофии является креатинурия, отражающая степень атрофии мышечной ткани; одновременно у больных снижается экскреция с мочой креатинина. Креатининовый показатель мочи, представляющий отношение креатинина к сумме креатина и креатинина и в норме равный единице, снижается до 0,8 (С. Н. Давиденков). Содержание в плазме крови и в моче у больных амиотрофией свободных аминокислот существенно не меняется. Наследственные амиотрофии следует дифференцировать с мышечными атрофиями (см. Атрофия мышечная). Прогноз неблагоприятный при ранних и быстротекущих формах амиотрофии — больные погибают в детском возрасте. При поздних и медленно текущих формах прогноз относительно благоприятный: больные благодаря сохранным мышечным группам могут компенсировать функцию пораженных мышц. Следует избегать переохлаждения и переутомления, ведущих к обострению заболевания.
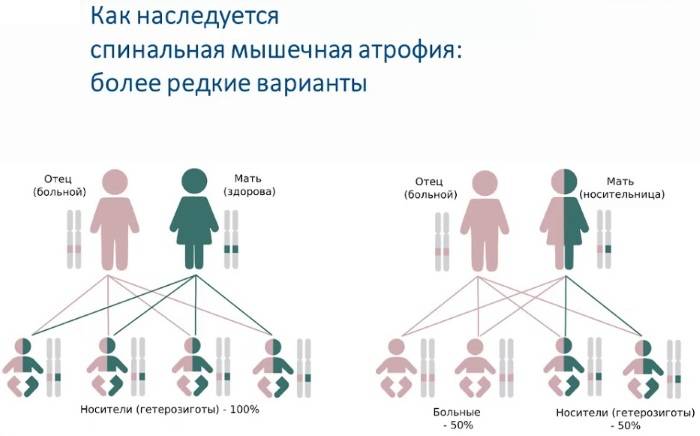
Родителям, имеющим ребенка, больного спинальной или невральной амиотрофией, рекомендуется воздержание от дальнейшего деторождения.
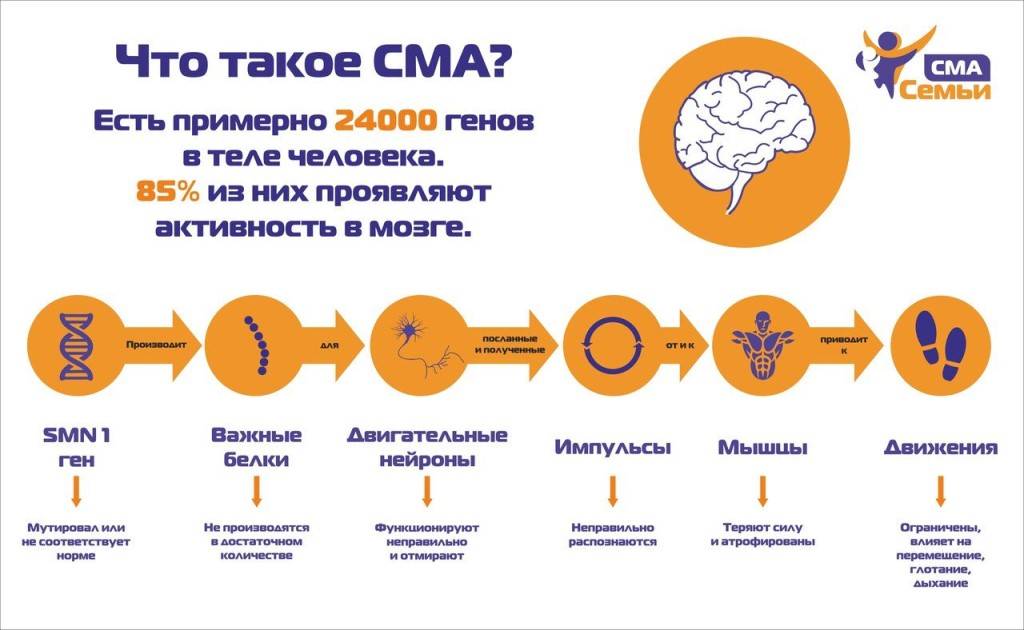
2.Причины
На клеточном уровне мышечная ткань устроена так, что ее длительное бездействие воспринимается организмом как повод для «сокращения штатов», т.е. для избавления от энергозатратных, но не используемых мышечных волокон. Поэтому наиболее распространенная причина атрофии бедренного квадрицепса – продолжительный период вынужденной неподвижности после травмы, масштабного хирургического вмешательства, комы и т.п.
Однако спектр возможной этиологии отнюдь не ограничивается сказанным. К такой атрофии могут приводить также врожденные, генетически обусловленные аномалии и дегенеративные заболевания мышечной ткани, аутоиммунные болезни, миозит (воспаление мышц), суставная патология, эндокринные и/или метаболические расстройства, а также дегенеративно-дистрофические процессы в проводниковых структурах нервной системы. Кроме того, атрофия может начаться в силу алиментарных причин, т.е. на фоне длительного и глубокого дефицита питательных веществ в связи с голоданием (в том числе и при применении экстремальных диет «для похудения»). Некоторые хронические и острые интоксикации также способны запустить атрофический процесс в мышцах. Наконец, атрофия может быть следствием естественного угасания метаболизма и активности в старческом возрасте.
Психотерапия
Начало клинических проявлений и последующие ухудшения, как правило, провоцируются психическим стрессом. Более того, тяжелое заболевание мышц, само по себе, серьезный психический стресс. На фоне болезни часто развиваются вторичные депрессии с апатией и нежеланием заниматься своим здоровьем. А это ведет к прогрессированию болезни и возможной гибели пациента.
Если это нужно, мы проводим нашим пациентам курс психотерапевтического лечения. Наши пациенты становятся более устойчивыми к психическим раздражителям, становятся активнее, позитивно относятся к лечению, и это незамедлительно сказывается на работе мышц.
Кроме того, мы обучаем пациентов специальным психотехникам для работы с собственными мышцами.
При наличии признаков депрессии возможно назначение современных антидепрессантов (селективные ингибиторы обратного захвата серотонина, такие как Ципралекс, Флуоксетин и др.). Эти антидепрессанты не снижают, а повышают активность пациента и настроение, не вызывают мышечной слабости.
«Терапию ждали очень давно»
— Как вы узнали о первых препаратах, помогающих при СМА?
— Разработкой генокоррекции и других видов патогенетического лечения в мире начали заниматься много лет назад. Мы также старались участвовать во всех заграничных конференциях по нервно-мышечным заболеваниям. И, конечно, такую терапию мы с пациентской организацией ждали очень давно.
О разработке первых препаратов мы узнали, когда у нас в областной больнице появился более-менее устойчивый интернет. До этого, в конце 90-х, читали актуальную иностранную литературу урывками, на ночных дежурствах. Доступ в интернет был через модем — как только находишь что-нибудь интересное, связь обрывалась.





Если интернет устойчивый и ты владеешь языком, то вся литература тебе доступна. Мне повезло — в 2001 году я ездила в командировку в Австралию, там была возможность подключиться к национальной американской библиотеке, где были собраны все публикации на интересующие меня темы.
— Как вы начали внедрять на базе института «Спинразу»?
— К сожалению, мы не участвовали в её клиническом исследовании, хотя производители нам делали соответствующий запрос. Дело в том, что у нас на тот момент было мало зарегистрированных пациентов со СМА I типа. Но нам удалось поучаствовать в программе дорегистрационного доступа к препарату, получить свой собственный опыт, потому что всё-таки есть разница — читать результаты клинических исследований или лично наблюдать пациентов. Мы убедились на собственном опыте, что «Спинраза» работает.
Мы также участвуем в клинических исследованиях «Эврисди». Они всё ещё продолжаются, но поскольку предварительные данные показали хороший результат, то препарат уже зарегистрирован. Тут мы тоже видим положительный эффект.
— А как вы стали применять «Золгенсму»? Как проходило обучение специалистов? Ведь оно пришлось на начало пандемии коронавирусной инфекции.
Также по теме

«Мы были будто бы во сне»: как живут дети со СМА после приёма самого дорогого лекарства в мире
В последний день февраля отмечается Международный день редких заболеваний. Спинальную мышечную атрофию иногда называют «самой частой…
— Мы, конечно, знали о «Золгенсме» из научной литературы, но, помимо этого, некоторые активные пациенты, у которых была финансовая возможность, ездили в США и получали там препарат, а потом возвращались к нам для наблюдения. Проблема в том, что, пока они собирали деньги, оформляли визу, проходили согласование с клиникой, уходило много времени. Я сама видела пациентов, которые в полгода были ещё в хорошем состоянии, но поехали туда уже в год с лишним, и к тому моменту много потеряли.
Потом началась пандемия, и пациенты, даже у которых были средства на лечение, выехать за границу уже не могли. Тогда мы обратились к производителям «Золгенсмы», в компанию Novartis, с просьбой организовать обучение специалистов, чтобы вводить препарат у нас в институте. Наше руководство эту идею поддержало.
Обучение проходило онлайн. Нам прислали материалы, требования, был однодневный тренинг с несколькими презентациями, мы задавали вопросы. Плюс подготовительный период, это две-три недели: смотрели, где в институте будем делать инъекцию, как работают наши инфузоматы, какую мы будем проводить седацию, которая может иногда понадобиться, чтобы качественно поставить катетер. Также приезжали представители компании, выясняли, соответствует ли институт условиям для введения лекарства: требования стандартные, как и для многих других биопрепаратов.
— Существовал какой-то отбор среди российских клиник, которые претендуют на право введения «Золгенсмы»?
— Претендентов было немного — не все спешат рисковать. А требования были такие: это должен быть медцентр, который имеет опыт работы с пациентами со СМА, знает, как их вести и как дополнительно оказывать им поддержку
Производители могут и отказать, ведь важно не только сделать сам укол, но и наблюдать за такими детьми, понимать особенности их организма, корректировать побочные действия, знать, как их после лечения восстанавливать. Желательно, чтобы был опыт клинических исследований в области нервно-мышечных заболеваний
Поэтому наш центр был первым, кто подал заявку. Но мы ввели препарат второму ребёнку в России, первыми инъекцию сделали в Национальном медицинском исследовательском центре имени В.А. Алмазова. Обучение мы прошли одновременно.
Бессилие
Для маленьких пациентов со спинальной мышечной атрофией переломным стал август 2019 года. У их родителей впервые появилась надежда сохранить своим больным детям жизнь. Тогда в России был зарегистрирован препарат «Спинраза» («Нусинерсен»), признанный эффективным для лечения этого заболевания. В других странах его начали регистрировать с 2016 года, российские пациенты несколько лет провели в ожидании. Но после выяснилось, что лекарство получат далеко не все.
Сколько в нашей стране сейчас живет людей со СМА, точно не известно, официального перечня нет. С 2016 года пациентов подсчитывает фонд «Семьи СМА». За это время удалось собрать информацию о 989 пациентах и 250–300 погибших. Теоретически (исходя из общемировой статистики заболеваемости) больных может быть около 2–3 тыс., считают в фонде.
Спинраза_4
 Фото: Global Look Press/Sebastian Gollnow
Фото: Global Look Press/Sebastian Gollnow
Распределение препарата относится к полномочиям регионов. Субъект обязан обеспечить нуждающихся в лекарстве, если оно рекомендовано по жизненным показаниям врачебной комиссией, пояснила «Известиям» директор фонда «Семьи СМА» Ольга Германенко. При этом только 30 регионов сделали единичные закупки для своих пациентов (в общей сложности не более чем на 100 человек), добавила она.






В январе 2020 года в Красноярске погиб ребенок не с самой тяжелой формой СМА. Полгода его мама боролась за получение препарата, но одержать победу не удалось, рассказала Ольга Германенко. По ее словам, проблема актуальна и в Москве.
По данным, приведенным московской прокуратурой в ответе одной из матерей больного ребенка (документ есть в распоряжении «Известий»), в столице живут 124 ребенка, имеющих диагноз «спинальная мышечная атрофия» (цифры ведомство приводит со ссылкой на департамент здравоохранения Москвы). Из них препаратом обеспечивают только 12 человек. В письме от 15 июня сказано, что надзорное ведомство направило в столичное правительство письмо с «предложением рассмотреть вопрос о принятии мер».
«Известия» обратились в депздрав Москвы с просьбой объяснить ситуацию.
По словам Ольги Германенко, некоторые субъекты пытаются помочь своим жителям со СМА. Единственный регион, который полностью решил вопрос, — Псковская область. Правда, там только один пациент. В каждом из 30 других субъектов, пытающихся выделить деньги на лекарство, более десятка таких больных.
Спинраза_3
 Фото: ТАСС/Юрий Смитюк
Фото: ТАСС/Юрий Смитюк
— Одна инъекция стоит почти 8 млн рублей. В первый год терапии пациенту нужно шесть инъекций, во второй — в два раза меньше, в каждый последующий — по три инъекции. Неготовность регионов обеспечить всех нуждающихся понятна, — отметила Ольга Германенко.
В Минздраве РФ «Известиям» пояснили, что на данный момент клинические рекомендации по лечению спинальной мышечной атрофии не утверждены. Однако «Спинраза» включена в их проект.
— С учетом указаний президента Российской Федерации в настоящее время прорабатываются механизмы обеспечения пациентов со СМА необходимыми лекарственными препаратами, — сообщили в пресс-службе ведомства.
3.Симптомы и диагностика
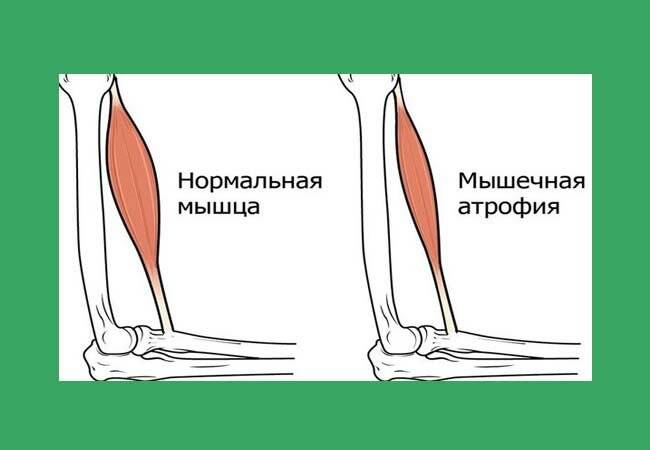
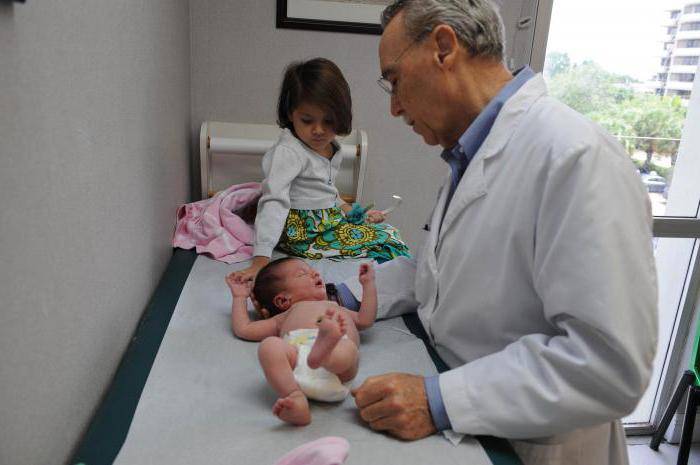
Клиническая картина атрофии квадрицепса характеризуется практически бессимптомным началом и медленным прогрессированием, порой многолетним – пока двигательные нарушения не вынудят пациента обратиться за помощью. Исключение составляют случаи, когда человек пытается резко встать (рассчитывая на привычный ему уровень физической активности) после длительного постельного режима: такая ситуация чревата неконтролируемым падением, вывихами, переломами и другими серьезными последствиями, поэтому в условиях стационара подготовка к смене режима всегда начинается заблаговременно.
Как правило, симптоматика нарастает постепенно, рано или поздно проявляясь видимым уменьшением мышцы в объеме, слабостью в ногах, изменениями («неуверенностью») походки. Со временем выраженность атрофических изменений возрастает, все больше усугубляя функциональную несостоятельность нижних конечностей.
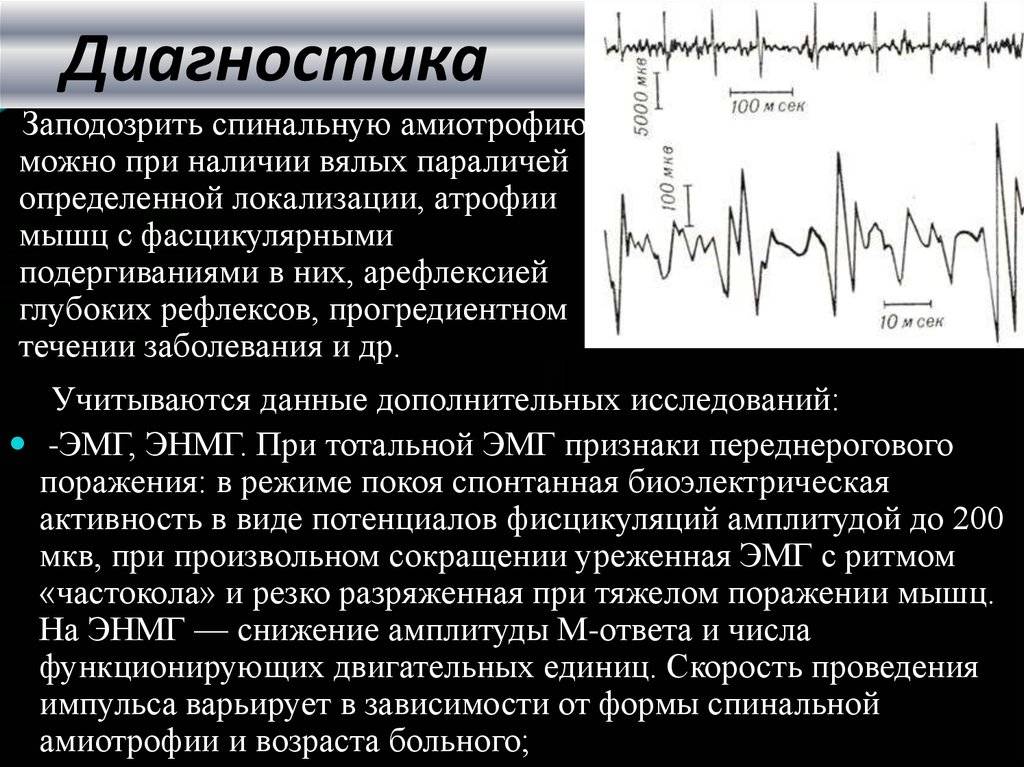
Наиболее информативным методом диагностики мышечной атрофии квадрицепса бедра, особенно ценным на ранних стадиях, является электромиография. Исследование нейромышечного реагирования позволяет выявить патологию еще на бессимптомном ее этапе. Большое диагностическое значение имеет также изучение анамнеза и динамики возникших нарушений.
Лечение спинальной мышечной атрофии
Ещё совсем недавно болезнь считалась неизлечимой и часто приводила к смерти или тяжёлой инвалидности. Все мероприятия были направлены на облегчение состояния пациента. В декабре 2016 года был разработан первый препарат Спинраза, который усиливает синтез белка SMN. Недостатком этого препарата является то, что он имеет высокую стоимость и лечение им нужно проводить на протяжении всей жизни больного СМА. Однако благодаря Спинразе были получены очень хорошие результаты в лечении и прогнозе у таких больных, особенно, если лечение начиналось на этапе доклинических проявлений. Дети в таком случае не отставали от своих здоровых сверстников по уровню физического развития.
В 2018 году был разработан препарат Золгенсма, который в мае 2019 года получил одобрение FDA. Он представляет собой генотерапевтическое лечение с заменой дефектного гена SMN1 на его нормальную копию. Переносчиком вектора AAV9 в клетку выступает инактивированный аденовирус. Итогом лечения становится нормальная выработка белка SMN. Человек получает полное исцеление после одной инъекции препарата. Дети, получившие Золгенсма на доклинической и ранней клинической стадии СМА, ничем не отличаются от своих здоровых сверстников. Даже пациенты, находящиеся на ИВЛ демонстрируют положительные результаты – начинают дышать самостоятельно, сидеть, вставать, ходить и т.д. Единственный недостаток препарата – это его стоимость. Он является самым дорогим на сегодняшний день препаратом в мире, 1 укол стоит более 2 млн.$. Активно ведётся разработка аналога Золгенсма в России и других странах мира, надеемся, что это произойдёт в ближайшем будущем, и многие семьи смогут получить спасительное лечение для своих детей.
Больным СМА также необходимо симптоматическое лечение по показаниям:
- метаболические препараты для улучшения функции митохондрий в мышечных тканях;
- противоэпилептические препараты – увеличивают образование белка SMN и улучшают клиническое течение болезни;
- муколитики и отхаркивающие – ввиду слабости дыхательной мускулатуры в лёгких скапливается мокрота, облегчить выведение которой помогут данные препараты;
- прокинетики и ИПП – в связи с нарушением глотания и снижением активности ЖКТ, а также частых ГЭРБ у больных СМА;
- гормональные препараты – при развитии сопутствующих осложнений;
- сахароснижающие лекарственные средства – в случае развития сахарного диабета на фоне СМА.
При развитии крайне выраженных контрактур суставов, деформаций грудной клетки и позвоночника рекомендованы ортопедические операции. Больным с постоянными рецидивами пневмоний ввиду аспирации пищи требуется проведение трахеостомии.
Лечение миопатии и амиотрофии – лекарства
Лекарства не излечивают дистрофию мышц, при этом они способны стимулировать регенерацию и рост мышечной ткани. В основном лекарственные препараты дают три эффекта:
- Улучшают питание мышц (L-карнитин, витамины, альфа-липоевая кислота, аминокислоты, вазоактивные препараты, биостимуляторы);
- Стимулируют рост мышц (анаболики);
- Уменьшают повреждение мембран клеток мышц (антиоксиданты).
Цели медикаментозного лечения:
- Повышение работоспособности мышц за счет увеличения количества полноценных мышечных волокон и снижения утомляемости;
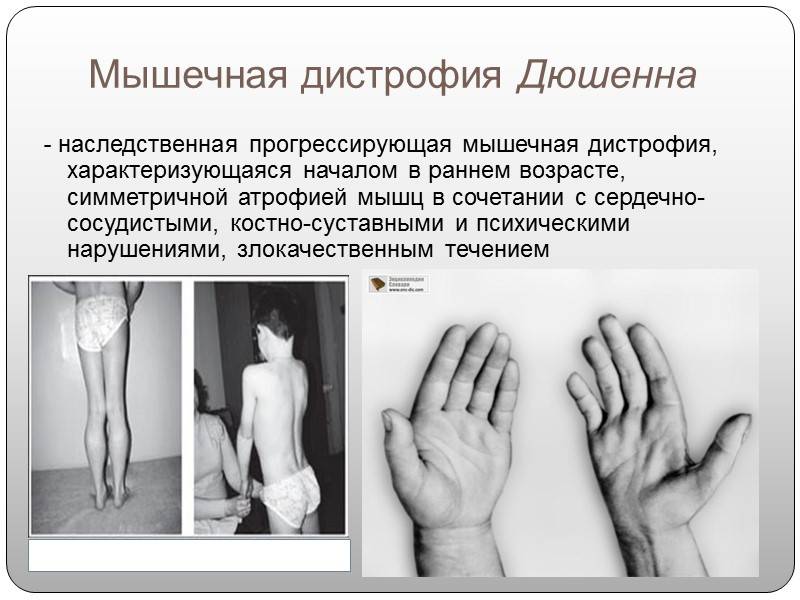
- Повышение сопротивляемости клеток мышечной ткани повреждению (установлено, например, что причиной миопатий Дюшенна и Беккера является генетически обусловленный дефект мембран мышечных клеток).
Универсального рецепта медикаментозного лечения при миопатии и амиотрофии не существует. Лечение приходится подбирать исходя из возраста, пола, типа миопатии, наличия сопутствующих заболеваний. Продолжительность курса – не менее трех месяцев.




«Нужна твёрдая рука»
— Как прошла первая инъекция?
— Конечно, мы волновались. Дело не в стоимости препарата (цены меня не впечатляют), а скорее в большой ответственности. Ведь надо аккуратно набрать в шприц конкретную дозу лекарства, потом ввести его за определённое время ребёнку (мы делаем катетеризацию сразу двух вен, чтобы за час прокапать нужный объём).
Боялись, что на любое внутривенное введение ребёнок может ответить какой-то немедленной аллергической реакцией. Но, к счастью, препарат хорошо переносится. По крайней мере, в период введения у нас не было никаких осложнений, как и в первые сутки после введения препарата.
Инъекция «Золгенсмы» вызывает повышение ферментов печени. В основном это проходит бессимптомно, дети себя хорошо чувствуют, тем более что печень — это орган, который прекрасно регенерируется.
Бывают и нестандартные случаи. Например, мы регистрировали девочку с одним весом, потом она выиграла в лотерею право доступа к препарату и набрала вес. А, как считается, чем старше ребёнок и чем больше он весит, тем выше риск, что повысятся ферменты печени. Но противопоказаний у девочки не было, препарат она уже выиграла, поэтому мы его ввели — и никаких негативных последствий не было, у неё всё хорошо.
— Не боялись, что ампула упадёт, разобьётся?
— Не разобьётся, там хорошие флаконы. И там их несколько, просто нужно их аккуратно набрать. Действительно, нужна сильная и твёрдая рука, потому что когда ты выводишь лекарство из флакона с вакуумом, то неминуемо система хочет его туда затянуть обратно, а ты должен её вытянуть и удержать. Потом так же другой флакон. А дальше уже дело техники, потому что введение идёт через специальное устройство с катетером.
А вот со «Спинразой» больше поводов для врача испытывать тревогу, потому что там один флакон. Ты его вскрыл, набрал, но не всегда получается сразу попасть в спинномозговой канал, а препарат после вскрытия можно хранить максимум три часа. Поэтому мы лекарство начинаем набирать только тогда, когда точно уверены, что никаких трудностей при введении не возникнет.
Получается, что «Золгенсму» вводить проще, но потом вести получивших инъекцию пациентов тяжелее, а сами они получают большую нагрузку.
— Но «Золгенсма» считается эффективнее и удобнее, ведь её достаточно ввести однократно, а «Спинразу» надо принимать пожизненно.
— Эффективность и удобство нам покажет только время. Официально существующие на данный момент препараты — «Золгенсма», «Спинраза», «Эврисди» — относятся к категории орфанных, они получили ускоренную регистрацию. Это была необходимая мера, учитывая, как протекает СМА I типа у пациентов и как меняется их жизнь после приёма.
Понятно, что пациентам кажется: чем больше возможностей использовано, тем лучше. Но на деле это не всегда так. Да, если начать лечить человека рано, то хороший эффект от всех препаратов, и тут, конечно, «Золгенсма» выгоднее, так как приём однократный, а не пожизненный. Но если симптомы СМА уже проявились, то пока нельзя сказать, какой метод лечения является преимущественным. Неврологический дефект всё равно может остаться, и тут важнее даже не само лечение, а восстановительная терапия и индивидуальный резерв пациента.
У меня мама хирург-ортопед, она всё удивляется — говорит, что раньше специалисты чётко знали: операция — это только половина дела, дальше надо реабилитировать пациента. А сейчас медицина интенсифицируется, операции происходят за день, потом человека нередко просто прекращают вести, не следят за его восстановлением.
— А вы поддерживаете связь с семьями пациентов? Может, чья-то история вам запомнилась больше всего?
— Со многими поддерживаю. Те, кто получил у нас лечение, каждую неделю или две отчитываются о самочувствии, приходят на осмотры. Но и тех, кто был у нас до появления «Спинразы» или «Золгенсмы», стараюсь не терять из виду.
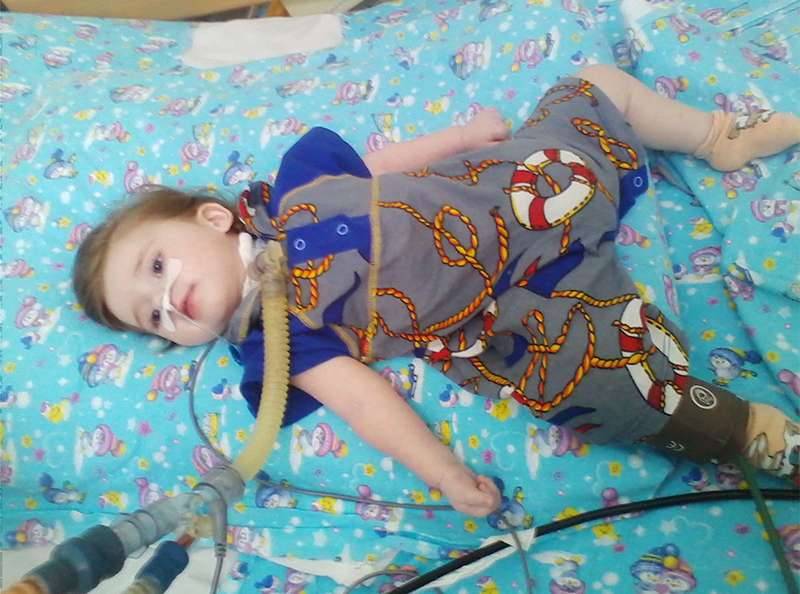
Я помню, сколько мы все бились за то, чтобы этот мальчик получил аппараты дыхательной поддержки, чтобы не отставала грудная клетка. И семья приложила максимум усилий, и со стороны врачей не было задержек, но патогенетического лечения на тот момент не существовало. Мальчик живёт, но качество жизни его и его близких значительно страдает. Поэтому большое счастье, что сейчас можно раньше начать терапию. Я думаю, что у нас не будет таких тяжёлых детей в дальнейшем.
Как диагностировать болезнь?
Появление характерных симптомов может указывать на развитие СМА, однако для подтверждения диагноза необходимо генетическое исследование. В 95% случаев заболевание связано с мутацией, представляющей собой полную или частичную делецию гена SMN1. Как правило, чтобы установить диагноз, достаточно выявить данную делецию4.





Для генетического тестирования обычно необходим образец крови.
Иногда проводится анализ крови на содержание креатинфоскиназы (КФК) —фермента, высвобождающегося из разрушающихся мышц. Однако этот тест неспецифичен, поскольку повышение уровня КФК свойственно не только СМА, но и многим другим нервно-мышечным заболеваниям, например, болезни Помпе.
В некоторых случаях может назначаться:
- Биопсия мышечного волокна;
- Электромиография, помогающая оценить уровень биоэлектрических потенциалов, которые возникают в мышцах.
Носителям делеции SMN1, которые могут передать мутацию по наследству, может быть рекомендована предимплантационная генетическая диагностика, используемая для скрининга пораженных СМА эмбрионов (при экстракорпоральном оплодотворении), а также пренатальное тестирование. Последнее включает анализ ворсин хориона, бесклеточный анализ ДНК плода и другие методики.
Екатерина Мартынова, мама Артёма
— Мы молодая семья из Воронежа, в мае мне будет 25 лет, а супругу 26. У нас было много планов, другие приоритеты. Но всё поменялось, когда Артёму, нашему единственному ребёнку, исполнилось 11 месяцев и мы узнали о его диагнозе. Речь идёт о начале 2019 года, тогда в мире была зарегистрирована только «Спинраза».
Мы записались в программу раннего доступа к «Спинразе» и получили таким образом шесть уколов. После первых четырёх инъекций Артём перестал слабеть, начали возвращаться навыки переворота на живот, на бок, стал громче кричать, начал сидеть. Курс закончили в мае 2020 года. Если бы мы не получили «Спинразу», то было бы очень страшно.
В мае 2019 в мире зарегистрировали «Золгенсму». Мы, конечно, осознавали, что такую сумму тем более невозможно собрать. Но потом стали встречать истории, когда это всё же удавалось: на Диму Тишунина, на Матвея Чепуштанова. И тоже решили попробовать.
Деньги собирали на протяжении полугода. До начала пандемии COVID-19 успели провести флешмоб, ярмарку на Масленицу, концерт в поддержку сына. Об Артёме говорили на всех городских праздниках, после этого сбор на сутки ускорялся.

Последние три месяца сбора мы сидели дома и работали через соцсети и звонки. Мы волновались, что всё затихнет на карантине, но на самом деле нам пандемия помогла: люди больше времени проводили в интернете и там узнавали об Артёме. Сбор мы закрыли своими силами, без перевода от какого-то одного человека, но нам помогали крупные организации. Например, один из банков перевёл около 12 млн.
Укол «Золгенсмы» Артём получил 30 июня 2020 года, в два года и два дня. Это не наша вина: дистрибьютора задержали с поставкой. Но врач в НИКИ педиатрии им. Вельтищева пошёл нам навстречу, всё-таки два дня — не критичная разница, тут важнее ориентироваться на вес ребёнка. Препарат ввели за час, первое время поднялась температура, но, как объяснили врачи, это обычное явление. Через две недели всё прошло, а через месяц анализы пришли в норму.
Сейчас Артёму два года и восемь месяцев. Он чувствует себя хорошо, крепнет, становится сильнее. Он, конечно, не побежал, не встал, но ползает с поднятой головой, отталкивается ручками и ножками, поднимает корпус. Это для кого-то мелочь, но для нас — прогресс семимильными шагами. «Золгенсма» работает, и это чудо. В Воронеже ходим в бассейн и на ЛФК, скоро полетим в Тюмень делать ортезы.
Наш сын — настоящий герой. Требовательный мальчик, целеустремлённый, шустрый, энергичный, хотя нам это пока не на руку, потому что концентрация на нуле, а нам нужно терпение. Он добрый, сильный, нежный, ласковый, харизматичный. Когда вырастет, будет непростым человеком, как мне кажется.
Болезнь Артёма сильно изменила нас с мужем. Поменялись приоритеты, мы начали понимать, что в этой жизни ценно. Эта ситуация сплотила нашу семью, мы стали больше друг друга ценить, между нами появилась особенная связь. Я сейчас получаю высшее образование, в этом году у меня диплом. На работу не вернусь, но ищу занятие, чтобы можно было совмещать с семьёй. Папа наш предприниматель, амбициозный, теперь у него ещё больше стимула работать. И, конечно, мы помогаем и продолжим помогать другим деткам. Необходимость тратить свои силы и энергию на это я особенно ясно поняла за время сбора. Чем больше отдаём, тем больше приходит
Важно делать это бескорыстно и не ждать чего-то взамен
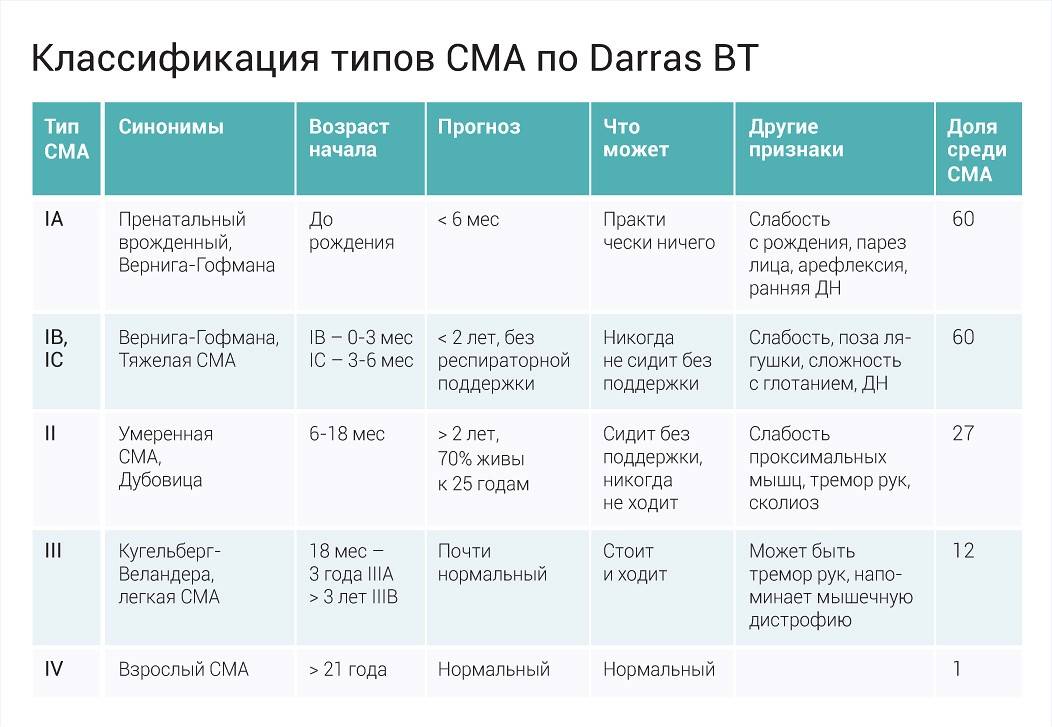
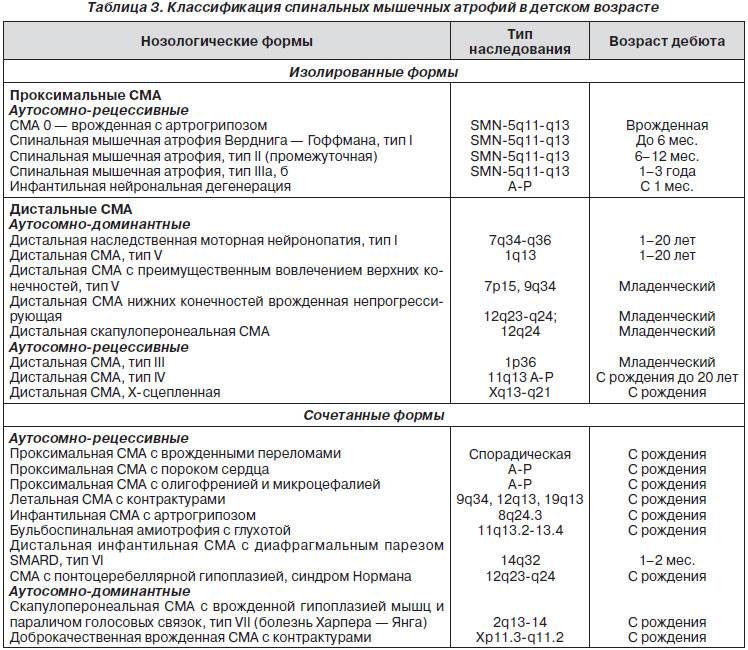
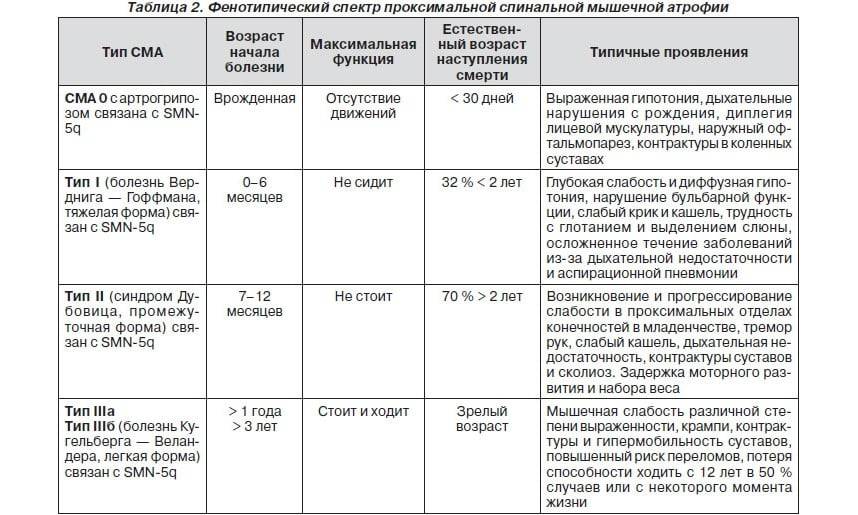
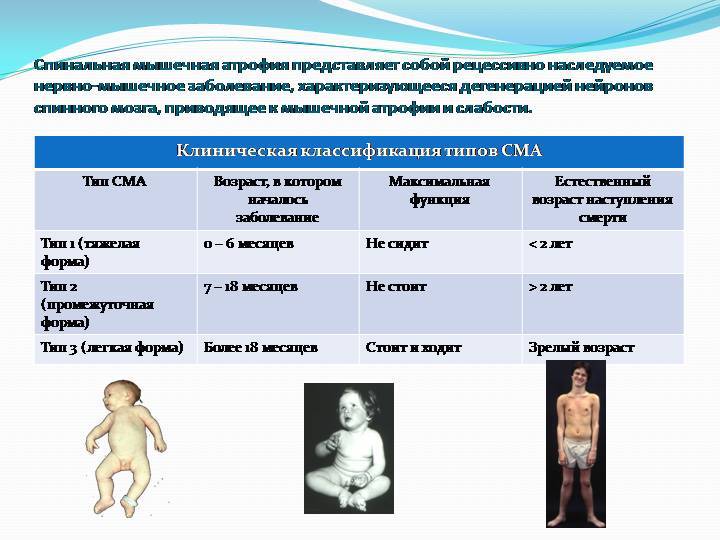
Клиническая картина и классификация СМА
Клиническая картина зависит от формы СМА. Все спинальные мышечные атрофии подразделяются на детские и взрослые. Детские в свою очередь также делятся на ранние, поздние и ювенильные.
 |
Детские спинальные мышечные атрофии:
- СМА 0;
- СМА I – болезнь Верднига-Хоффмана;
- СМА II – болезнь Дубовица;
- СМА III – болезнь Кюгельберга-Веландер.
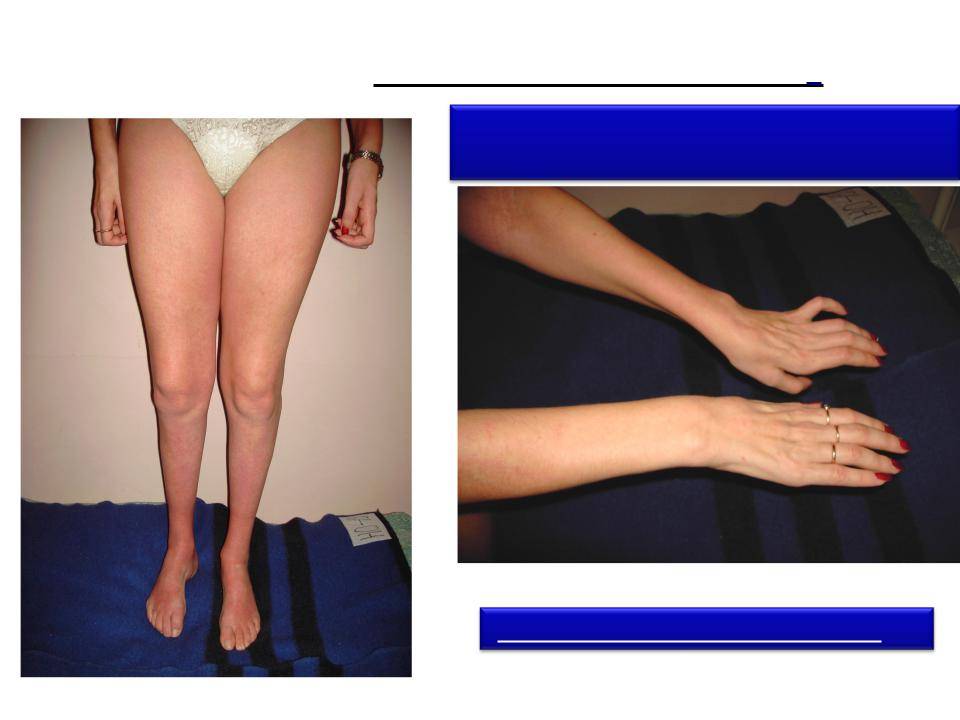
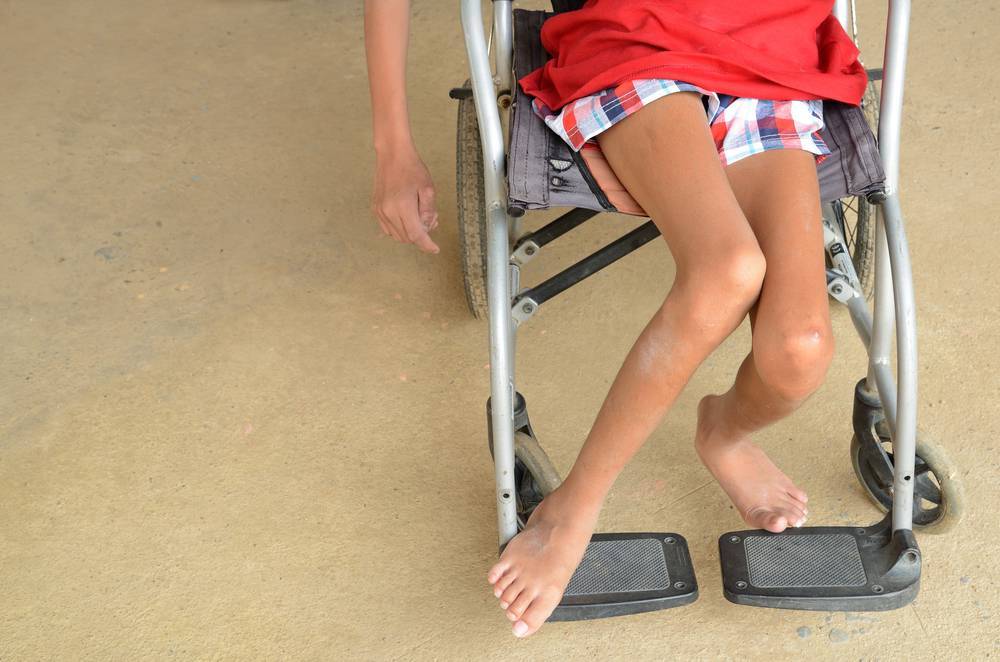
Самым неблагоприятным считается нулевой и первый тип СМА. При нулевом типе дети рождаются уже с контрактурами суставов и как правило сами не могут дышать в связи с поражением дыхательных мышц, их сразу подключают к ИВЛ. Прогноз неблагоприятный. При первом типе клиника начинается в промежутке с первых дней жизни и до 6 месяцев. Дети имеют трудности с сосанием, глотанием, дыханием, не держат голову, не могут сидеть. Клиническая картина СМА II проявляется в возрасте 7-18 месяцев, дети, больные этой формой не могут ходить самостоятельно и стоять, хотя все остальные функции (глотание, сосание, способность сидеть, держать голову, поворачиваться) у них сохранены. Прогноз в этом случае будет зависеть от степени вовлечения в патологический процесс дыхательной мускулатуры. Третья форма – болезнь Кюгельберга-Веландер является самой благоприятной СМА детского возраста. Клиника начинается после 1.5 лет. Пациент может стоять, но испытывает сильную слабость и вынужден передвигаться в коляске.





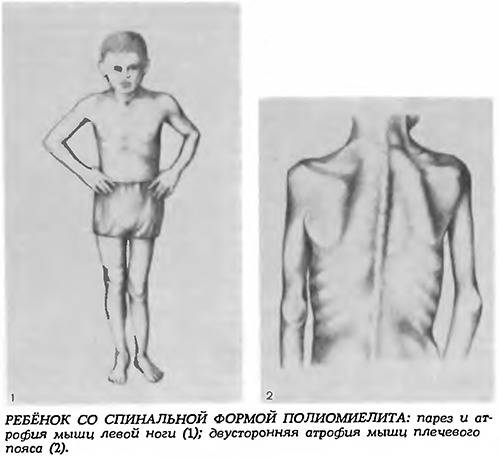



Взрослый IV тип СМА встречается у людей старше 35 лет и особо не влияет на продолжительность их жизни. Клиническая картина сопровождается снижением периферических сухожильных рефлексов, слабостью мускулатуры, фасцикуляциями и нарушением ходьбы. Помимо основных 4-х форм СМА встречаются дистальные, изолированные и сочетанные формы и синдромы. Изолированные формы представляют собой поражение только спинальных мотонейронов. Сочетанные комбинируются с врождёнными пороками сердца, олигофренией, глухотой и другой патологией.
Промежуточные данные 3-й фазы STR1VEпо состоянию на 27 сентября 2018 г. (дата среза данных для анализа)
STR1VE — это продолжающееся открытое простое многоцентровое исследование с однократной дозой, предназначенное для оценки эффективности и безопасности однократного внутривенного введения Zolgensmaу пациентов с СМА 1-го типа, которые на момент генной терапии не достигли возраста шести месяцев. Исследование было разработано для охвата как можно более широкой популяции пациентов с СМА 1-го типа с одной или двумя копиями резервного гена SMN2, которые имеют биаллельную делецию гена SMN1 или точечные мутации. Эти критерии хорошо соответствуют популяции пациентов, которые были зарегистрированы в основном испытании 1-й фазы START, при этом потенциально предоставляя лечение некоторым из более редких субпопуляций на исследовательской основе. Исследование STR1VEпланируется завершить в 2020 году.
По состоянию на 27 сентября 2018 года 21 из 22 (95 процентов) пациентов были бессобытийно живы (прим. «событие» смерть или необходимость в длительной вентиляции легких более 16 часов в сутки). Средний возраст составлял 9,5 месяцев, 6 из 7 (86 процентов) пациентов смогли достичь возраста в 10,5 месяцов или старше без событий на указанную дату. Естественная история развития заболевания без лечения показывает, что 50 процентов детей с СМА 1-го типа не выживут или будут нуждаться в постоянной вентиляции к возрасту 10,5 месяцев.
Оценки по шкале специального теста нервно-мышечных расстройств CHOP-INTEND увеличились в среднем на 7,0 баллов через месяц после применения терапии и на 11,8 баллов через три месяца, отражая улучшение двигательной функции по сравнению с исходным уровнем.
Эти данные сопоставимы с улучшениями CHOP-INTENDпри предложенной терапевтической дозе когорты (2-я когорта) в основном исследовании START, которое продемонстрировало среднее соответственное увеличение на 9,8 и 15,4 баллов на том же отрезке времени. Раннее увеличение баллов по CHOP-INTENDможет быть связано с возможным достижением моторных навыков пациентами.
Предварительные оценки пациентов, получавших Zolgensma, показали улучшение двигательных навыков. Три пациента могли сидеть без поддержки в течение не менее 30 секунд по состоянию на 27 сентября 2018 года (медиана возраста — 9,4 месяца) и восемь пациентов достигли того же рубежа к 31 декабря 2018 года (средний возраст — 12,5 месяцев).
| контрольная отметка, кол-во пациентов n (%) | 3 фаза исследования STR1VE, n=22 | |
| 27 сентября 2018 | 31 декабря 2018 | |
| Держит голову прямо 3 и более секунд без поддержки | 12 (54,5) | 17 (77,3) |
| Переворачивается со спины и в правую, и в левую стороны | 3 (13,6) | 7 (31,8) |
| Сидит без поддержки 30 и более секунд | 3 (13,6) | 8 (36,4) |
| Стоит с помощью | 1 (4,5) | |
| Средняя продолжительность наблюдения при сборе данных | 5,5 месяцев | 8,1 месяцев |
| Средний возраст при сборе даных | 9,4 месяцев | 12,5 месяцев |
| Пациенты старше 12 месяцев, кол-во n (%) | 5 (22,7) | 13 (59,1) |
Наблюдения за безопасностью сравнимы с наблюдениями в 1-й фазе ключевого исследования START. Неблагоприятные события, представляющие особый интерес, включая повышение уровня трансаминаз, снижение количества тромбоцитов и тромбоцитопения, были преходящими и не вызывали каких-либо долгосрочных последствий. Один пациент умер от дыхательной недостаточности, которая была признана исследователем и независимым советом по мониторингу безопасности данных не связанной с лечением. Этот пациент продемонстрировал значительное улучшение моторики, с увеличением CHOP-INTENDна 27 баллов по сравнению с исходным уровнем через пять месяцев после вливания.
AveXis благодарен мужественным пациентам и семьям, которые принимают участие в наших испытаниях, что позволяет нам продолжать прилагать усилия по осуществлению значительных изменений в жизнь пациентов с редкими генетические заболеваниями.
Диагноз – миопатия (амиотрофия). Что делать?
Сегодня панацеи от наследственных мышечных дистрофий не существует. Однако правильное лечение позволяет ощутимо затормозить атрофию мышц и увеличить регенерацию и рост новой мышечной ткани, и даже вернуть некоторые утраченные возможности. Лечение миопатии и амиотрофии требует повседневного выполнения ряда медицинских процедур, поэтому мы не только оказываем лечебную помощь, но и обучаем родственников пациентов и/или самих пациентов самостоятельному выполнению необходимых процедур.
Лечение в нашей клинике включает в себя следующее:
- Прием лекарств по схеме, которая расписывается на срок 3-12 месяцев;
- Лечебное питание;
- Физиотерапия;
- Массаж;
- Гимнастика;
- Психотерапия и духовные практики;
- Нейропсихологическое развитие (для детей, отстающих в интеллектуальном развитии).
По каждому из перечисленных пунктов проводится подробное обучение. Мы настаиваем на обучении самостоятельному выполнению процедур, т.к. стремимся сделать регулярное и достаточное по объему лечение еще и доступным и дешевым.